

Abstract
Many aggressive hematologic malignancies are exquisitely responsive to immunotherapy. While allogeneic (allo) hematopoietic stem cell transplantation (SCT) is the prime example of anti-leukemia immunotherapy, it is highly toxic, limiting its application to patients with aggressive disease and a good performance status. Although targeting leukemia-associated antigens (LAA) with antigen specific cytotoxic T lymphocytes (CTL) and chimeric antigen receptor (CAR) T cells has proven to be a successful immunotherapeutic option, the immune response generated with these approaches often fails to fully eradicate the underlying malignancy. The expression of immune inhibitory molecules and their ligands by myeloid leukemia cells and leukemia-specific T cells could contribute to the failure of cellular immunotherapy approaches for acute myeloid leukemia (AML). Specifically, blocking the PD1/PD-L1 pathway using antibodies was shown to enhance the graft versus leukemia response in murine leukemia transplant models, and promising results with immune checkpoint blockade have been demonstrated in clinical trials for patients with AML. We therefore investigated the role of blocking the PD1/PD-L1 pathway in combination with adoptive cellular immunotherapy in the setting of AML. We used pembrolizumab in combination with CTLs that target the HLA-A2 restricted myeloid leukemia antigens CG1, derived from cathepsin G, and PR1, derived from neutrophil elastase and proteinase 3.
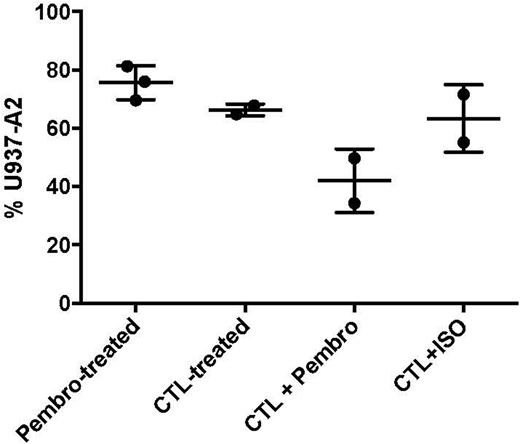
Using a standard calcein AM in vitro cytotoxicity assay, we co-cultured AML targets, including U937 HLA-A2+ (U937-A2) AML cell line and primary patient AML, with CG1-CTL and PR1-CTL. AML cells were loaded with calcein AM and then incubated with CTL at increasing effector to target ratios. Pembrolizumab or isotype antibody was added to the cultures. After 4 hours, calcein AM was measured to determine cell viability. Our results demonstrated that U937-A2 cells and patient primary AML samples can be effectively lysed by CG1/PR1 CTL. In vitro lysis of AML by CTL was enhanced after the addition of pembrolizumab in comparison with the AML cells that were co-cultured only with CTL. Killing was dose-dependent and decreased at the lower effector:target ratios. We then tested whether anti-leukemia activity can be enhanced in vivo. For these experiments, NOD/SCID gamma (NSG) mice were engrafted with U937-A2 cells or patient AML intravenously (IV) via tail vein at a dose of 1 x 106 to 1 x 107 cells. After confirming engraftment (1-5% PB HLA-A2+/human (h) CD45+ cells), CG1-CTL and PR1-CTL (0.5 x 106) were administered to mice IV via tail vein and pembrolizumab or isotype antibody [100ug/mouse] was given intraperitoneally 4 times over 2 weeks. Mice were monitored for clinical graft versus host disease (GVHD) and AML 3 times/week. Mice were sacrificed at approximately 2 weeks following treatment and bone marrow (BM) was processed and analyzed for residual AML (Human[h] CD45+/mouse[m] CD45- cells) by flow cytometry. Our data demonstrate a decrease in U937-A2 (Figure) and primary AML after treatment with CG1/PR1-CTL. This decrease was enhanced when pembrolizumab was administered to the CTL-treated mice in comparison with mice treated with pembrolizumab only, CTL only, or CTL+ isotype. Furthermore, there was no increased toxicity or GVHD after the addition of pembrolizumab. Together these data highlight the potential for combination immune checkpoint blockade and antigen specific CTL to eradicate AML in vitro and in vivo.
Daver:Incyte: Consultancy; BMS: Research Funding; ImmunoGen: Consultancy; ARIAD: Research Funding; Sunesis: Consultancy; Alexion: Consultancy; Novartis: Research Funding; Daiichi-Sankyo: Research Funding; Sunesis: Research Funding; Incyte: Research Funding; Karyopharm: Research Funding; Otsuka: Consultancy; Novartis: Consultancy; Kiromic: Research Funding; Karyopharm: Consultancy; Pfizer: Consultancy; Pfizer: Research Funding.
This icon denotes a clinically relevant abstract

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal